| 文献分享 | 您所在的位置:网站首页 › scrna seq怎么读 › 文献分享 |
文献分享
|
在对scRNA-seq的数据做常规流程分析的时候,最重要的一步就是给定细胞亚群的名字,那么这时候所用的方法就很关键。现在人们普遍使用的注释方法有手动注释、R包注释。但是不管怎么注释,都是离不开marker gene的。所以我今天想跟大家分析一下这篇文献,是关于在做scRNA-seq的细胞亚群注释时,所用的挑选marker gene的方法比较。 然后在往推文上截图的时候发现,图上字太小了,清晰度不够。对这篇文献感兴趣的可以去搜索下载下来看一看。 标记基因(marker gene)的概念与差异表达(DE)基因的概念密切相关,但两个概念并不是不同义词。严格来说,标记基因选择是DE基因鉴定的一个子集,有效且有用的标记基因具有并非所有DE基因所共有的特定特征。广义上,我们将标记基因定义为可用于区分细胞亚群的基因。 文献标题及背景A comparison of marker gene selection methods for single‑cell RNA sequencing data.  背景:分析 scRNA-seq 数据的一个常见步骤是选择所谓的标记基因,最常见的目的是对样本中存在的生物细胞类型进行注释。本文是对scRNA-seq 数据中选择标记基因的 59 种计算方法进行了基准测试。 在本文中,我们比较了 59 种在 scRNA-seq 数据中选择标记基因的方法。这些方法使用 14 个 scRNA-seq 数据集(包括一系列协议和生物样本)以及另外 170 多个模拟数据集进行了基准测试。对各种方法(应用传统统计方法和现代机器学习方法)进行了基准测试,这些方法是专门为标记基因选择任务而设计的,不适用于更一般的 DE 基因检测。具体来说,本文比较了各种方法恢复模拟标记基因和专家注释标记基因的能力、预测性能和所选基因集的特征、内存使用率和速度以及实施质量。 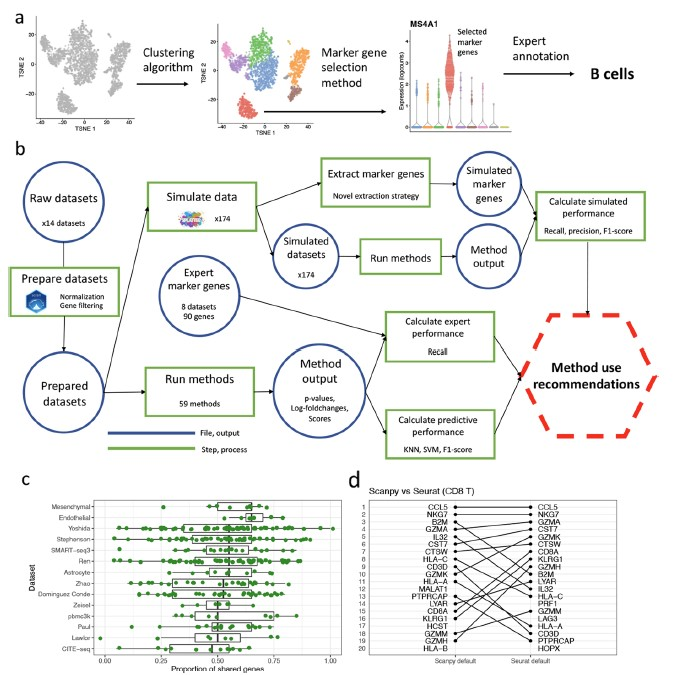 屏幕截图2024.03.06_13.40.02.png 本文的研究表明,尽管大多数方法表现良好,但所选标记基因的质量仍有差异。值得注意的是,较新的方法不能全面优于较旧的方法。这些案例研究强调,即使是在相似的方法之间,也存在很大但未得到重视的方法学差异,这可能在某些情况下对它们的产出产生很大影响。 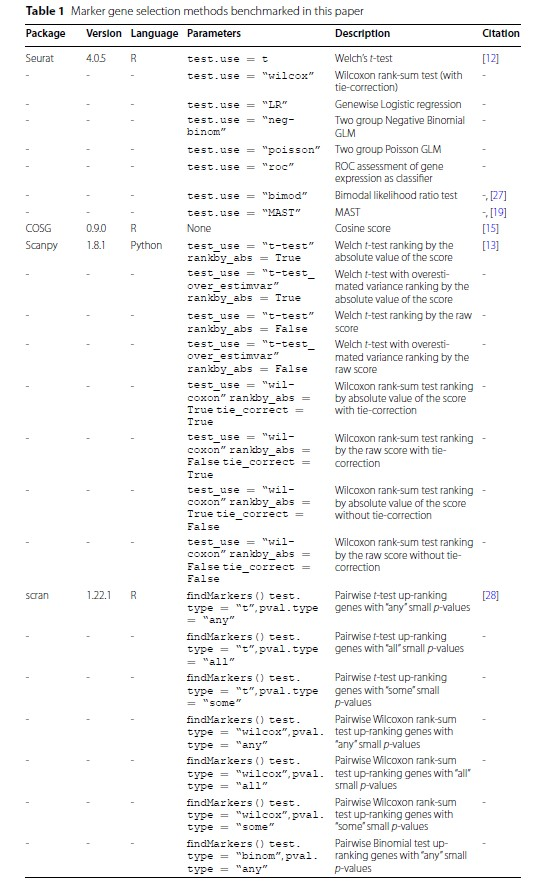  文章在14个真实数据集(表2)和170多个额外的模拟数据集上对59种方法(表1)的性能、计算效率和输出特征进行了基准测试。文章选择了真实数据集来代表一系列方案(包括10X Chromium、Smart-seq3、CITE-seq和mars -seq)和大小范围(从约3,000个细胞到60,000个细胞)。  方法一致性和输出特性。 方法一致性和输出特性。a基于所选择的最多前20个基因中共享基因的比例的方法层次聚类的树状图表示。方法由实施它们的包装来标记,它们是只选择上调的标记基因还是同时选择上调和下调的标记基因,以及它们输出的是一组标记基因还是根据标记基因状态的强度排列的基因。b多种特征汇总(平均数据集)方法选择的最多前5个标记基因的特征。方法按字母顺序排序。c 3个特征(曲线下面积、对数变化倍数和Cohen’s d)汇总了方法选择的最多5个标记基因的1 -vs-rest效应大小(在数据集上取平均值)。对这三个特征进行分位数标准化,并根据不同数据集的中位数评分对方法进行排序 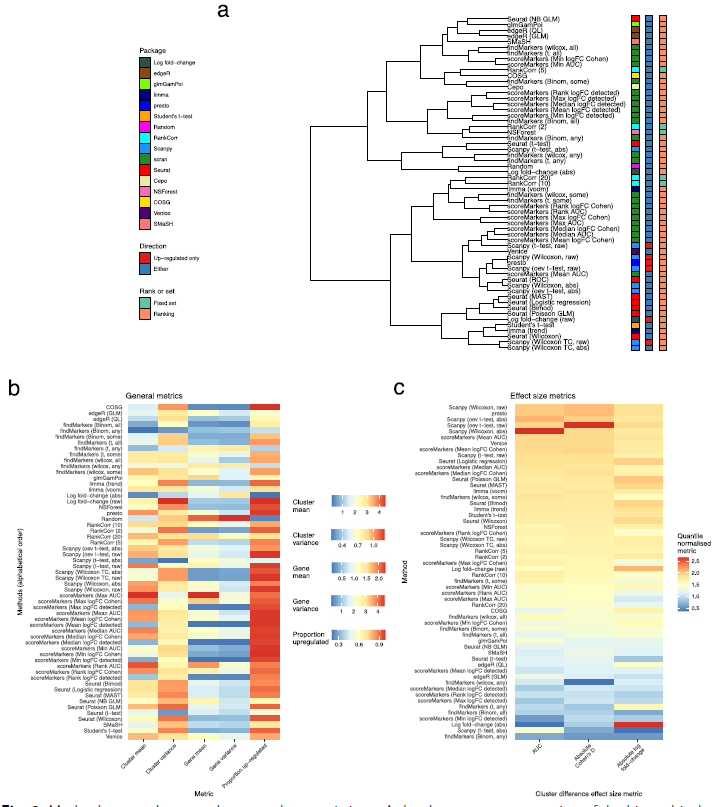 使用模拟数据集的方法比较。 使用模拟数据集的方法比较。a基于所有真实数据集的模拟场景下所有方法的计算召回率。根据仿真模型参数选择的标记基因作为基础真值。方法在热图中按场景的中位数召回率从上到下排列。b和(a)一样,但现在更精确了。c与(b)中相同,但计算F1分数。这些结果在模拟集群和模拟重复上平均,并在飞溅模拟模型3中选择了20个基因和DE因子的位置参数。 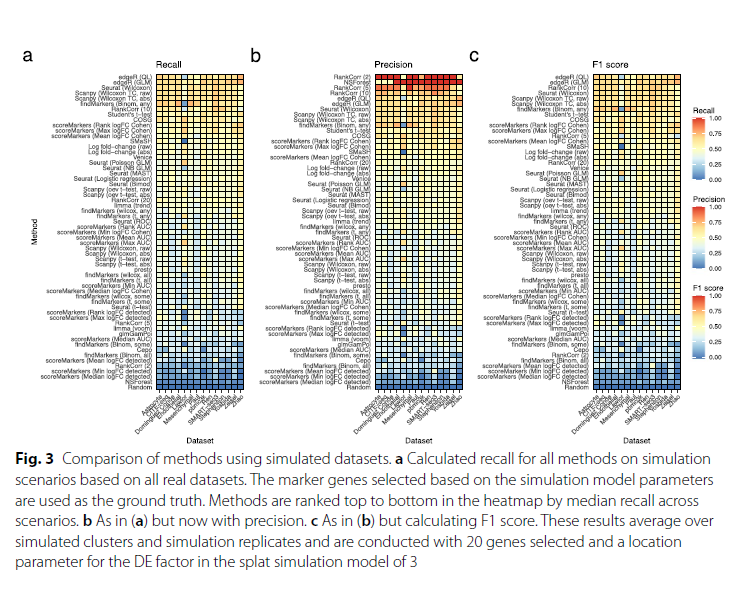 基于专家注释标记基因的方法比较。 基于专家注释标记基因的方法比较。回顾在Lawlor数据集中选择标记基因的方法,使用一组专家注释的标记基因作为 ground truth。使用描述Lawlor数据集的原始出版物中用于标注聚类的标记基因作为专家标注的标记基因集。从每种方法的输出结果中选择最优基因。b如(a),但对于pbmc3k数据集,使用(最多)来自每种方法的前10个标记基因,并将专家注释的标记基因集作为Seurat包的“引导聚类教程”中使用的标记基因。c使用Lawlor数据集中所选标记基因成功注释的集群数量(其他细节见a)。如果所选标记基因包括该集群的所有专家注释标记基因,则将特定集群定义为成功注释。d注释分析的成功与(c)相同,但对于pbmc3k数据集,专家注释的标记基因的细节和选择的标记基因的数量如(b) 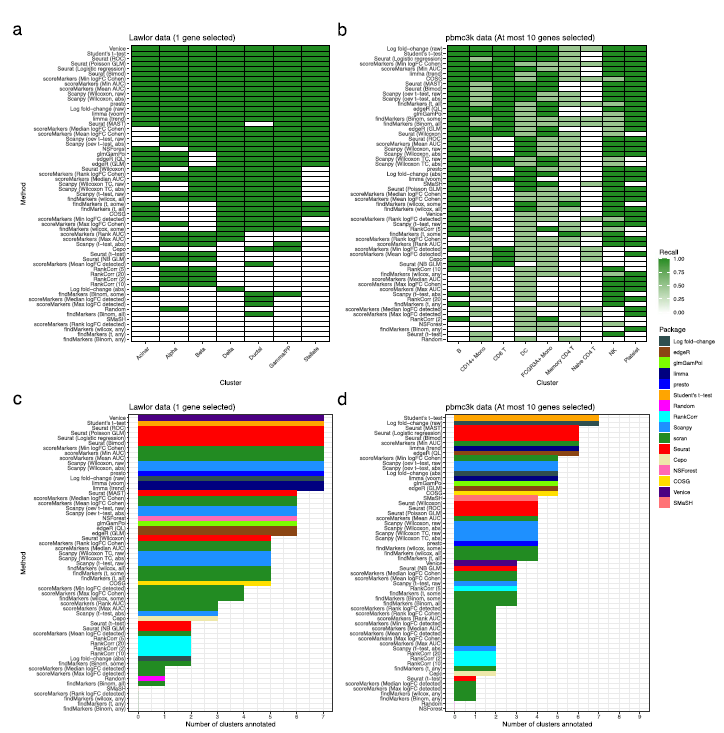 使用预测性能的方法比较。 使用预测性能的方法比较。使用Seurat Wilcoxon方法在pbmc3k数据集中选择的基因集,用混淆矩阵表示KNN(三个最近邻)分类器的性能。使用Zhao数据集中所有标记基因选择方法所选基因的KNN分类器F1评分的中位数。每一分都是5次中的F1得分。每个数据集KNN分类器F1评分中位数(跨折线平均)的z-score。方法根据它们在不同数据集中的平均z-score从上到下排序。 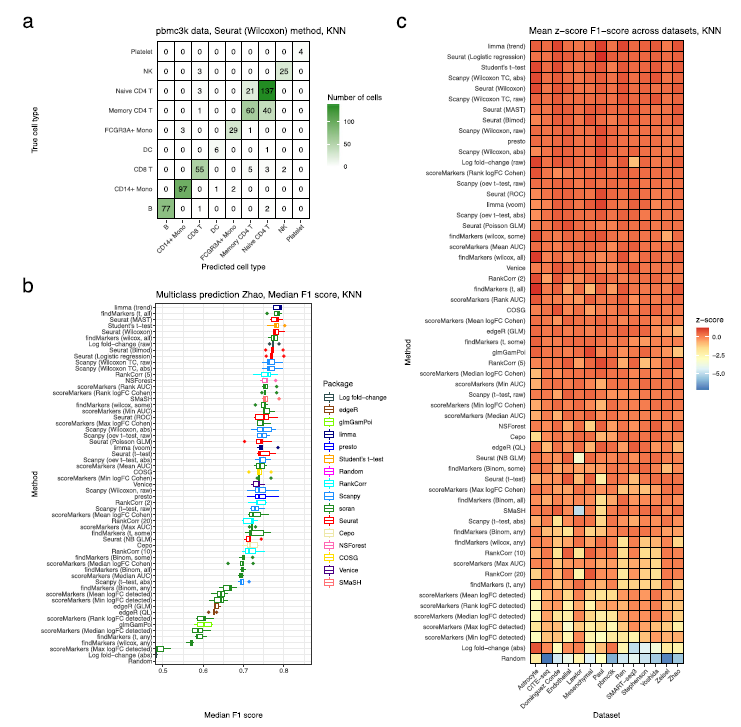 结果 结果使用 14 个真实 scRNA-seq 数据集和 170 多个额外的模拟数据集比较了这些方法的性能。比较了各种方法恢复模拟和专家注释标记基因的能力、预测性能和所选基因集的特征、内存使用率和速度以及实现质量。此外,还利用各种案例研究对最常用的方法进行了仔细研究,突出了其中存在的问题和不一致之处。结果凸显了简单方法的功效,尤其是 Wilcoxon 秩和检验、Student t 检验和逻辑回归、 特别是 Wilcoxon 秩和检验、Student t 检验和逻辑回归。 |
【本文地址】